 español
español inglés
inglés francés
francés alemán
alemánmutante sin cura


Las personas con enfermedad de
Huntington pueden padecer trastornos:

Musculares:
anomalías en la forma de caminar, aumento de la actividad muscular, espasmos musculares, movimientos involuntarios, problemas de coordinación, pérdida de músculo, dificultad para hablar y temblores.

Cognitivos:
lentitud para realizar actividades, amnesia, confusión, dificultad para pensar y comprender, falta de concentración o pérdida de la memoria.

De comportamiento:
comportamiento compulsivo, falta de autocontrol, irritabilidad o moverse nerviosamente.

Psicológicos:
alucinación, delirio, depresión o paranoia.

Estado de ánimo:
altibajos emocionales, ansiedad o apatía
Síntomas
La eventual muerte de neuronas en estas áreas cerebrales, involucradas en el control del movimiento y de los procesos cognitivos, son la causa fisiopatológica de los signos y síntomas observados en los pacientes con EH, los cuales en general consisten en movimientos involuntarios continuos (corea) que se hacen progresivos a medida que evoluciona la enfermedad. Además, los pacientes con EH muestran numerosos síntomas psiquiátricos, incluyendo depresión, ansiedad y, en algunos casos, demencia.
“La EH es un padecimiento severo tanto para el paciente como para aquellos que cumplen la función de cuidadores. La enfermedad suele presentarse entre los 30 y los 50 años de edad, aunque esto dependerá de la longitud de las tripletas descritas. A medida que la enfermedad sigue su curso, el paciente presenta movimientos más pronunciados e incontrolables, lo que termina incapacitando al paciente, culminando con su total dependencia y eventual muerte”, argumenta el profesor.
Estudio
Infortunadamente, no existe cura para la EH. De hecho, el único tratamiento (tetrabenazina) aprobado por la FDA y el Invima mitiga algunos síntomas de la enfermedad de manera parcial pero no modifica su curso. Esto se debe a que, a pesar de haber descubierto la mutación causal de la EH hace casi tres décadas, aún no se conoce la función de la proteína huntingtina normal, ni cuáles son sus potenciales efectores moleculares.
Esto quiere decir que, a diferencia de otras enfermedades como el Alzhéimer o Párkinson, en EH sabemos cuál es la alteración genética que causa la enfermedad, permitiendo a científicos estudiar el cerebro de personas con Huntington incluso antes de que se manifiesten los primeros síntomas. Esto les ha permitido estudiar lo que pasa en el cerebro durante el proceso de neurodegeneración, como si en vez de descifrar un asesinato con base en las pruebas de la escena del crimen, siguen al asesino (mutación) y a la víctima (neuronas) antes de que ocurra el crimen.
Durante los estudios de doctorado del profesor Luis Carlos Morales, realizados entre 2012 y 2018 en la Universidad de Alberta (Canadá), él y un equipo de investigadores descubrió que cerebros de modelos animales y células obtenidas de pacientes con EH, contenían una cantidad inferior de un lípido complejo conocido como gangliósido GM1, en comparación con células y cerebros sin la enfermedad. Este lípido se encuentra normalmente enriquecido en estructuras cerebrales tales como los axones neuronales y la cubierta de mielina, favoreciendo la supervivencia neuronal, la transmisión de señales y el transporte de moléculas en la superficie de la neurona. De manera notable, el suministro in vitro de gangliósido GM1 a neuronas expresando el gen HTT mutante, logró disminuir la mortalidad neuronal y disminuyó drásticamente la acumulación de proteína mutante.
“Tras encontrar resultados positivos en modelos in vitro de la EH, realizamos experimentos en animales portadores de la mutación de HTT, donde suministrábamos gangliósido GM1 directamente en el cerebro de los animales enfermos. Encontramos que esta terapia disminuye de manera dramática los signos motores y cognitivos de la enfermedad, así como incrementa la supervivencia de los ratones mutantes”, indica el experto.
A nivel histopatológico (examen microscópico de tejido) también encontraron que el tratamiento con gangliósido GM1 resulta en una mayor población neuronal, disminución de la inflamación cerebral y una mayor conservación de la mielina. Estos hallazgos son un gran paso adelante en la búsqueda de terapias enfocadas en la modificación de la progresión de la enfermedad.
Hasta la fecha, no existe ninguna cura e investigadores en todo el mundo tratan de hallar el tratamiento o fármaco que evite los estragos de la enfermedad de Huntington, que es un trastorno genético que se transfiere con un patrón mendeliano de tipo autosómico dominante: si uno de los padres padece la enfermedad, cada uno de sus hijos tendrá un 50% de probabilidad de desarrollarla.
Relacionados
Una mirada a la medicina del futuro
Edición 004 - intellecta
Los misterios del sida que descifran las bacterias del intestino
Edición 002 - intellecta
Implicaciones del estudio genético a debate en tertulia del HUN
Publicado en: lun, 27 ago 2018 05:05:00 - Grupo Prensa
Compartir artículo en:


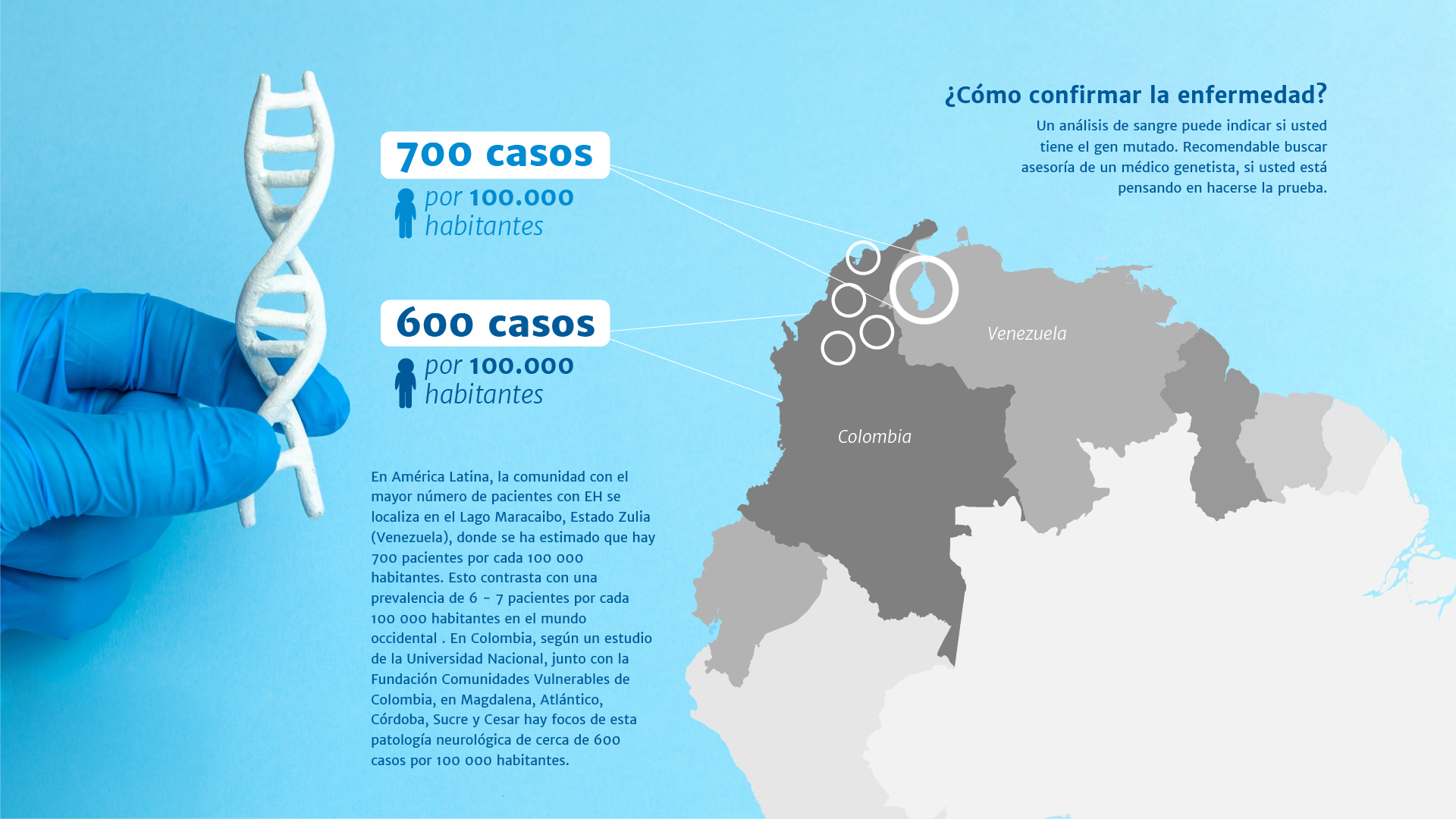
Latinoamérica
En América Latina, debido al aislamiento y al mecanismo de herencia de la enfermedad, existen comunidades en las que la prevalencia de la EH es muy superior al promedio global. La comunidad con el mayor número de pacientes con EH se localiza en el Lago Maracaibo, Estado Zulia (Venezuela), donde se ha estimado que hay 700 pacientes por cada 100 000 habitantes. Esto contrasta con una prevalencia de 6 - 7 pacientes por cada 100 000 habitantes en el mundo occidental.
En Colombia dos poblaciones parecen tener el mayor número de pacientes: Juan de Acosta (Atlántico) y El Difícil (Magdalena). Sin embargo, el profesor advierte que no existen cifras oficiales de pacientes y/o portadores (pacientes asintomáticos) del gen mutante, debido, en gran parte, a que nuestro sistema de salud no incluye el examen genético en el plan obligatorio de salud y a que el Estado no ha hecho mayores esfuerzos en conocer el estado de salud de estos pacientes en particular.
“En resumen, tenemos un total desconocimiento de quienes padecen EH o quienes portan el gen mutante sin presentar síntomas aún. La precaria caracterización de nuestra población con EH nos hace virtualmente invisibles ante el mundo y los adelantos farmacológicos y tecnológicos que se desarrollen en el futuro”, puntualizó el investigador.